|
Местный
Регистрация: 12.03.2018
Сообщений: 246
Спасибо: 0
Спасибо 6 в 5 постах
Репутация: 10
|
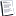
Таким образом, увеличение клеточного отношения AMP / ATP вызывает прогрессирующую активацию AMPK. ADP также может регулировать активацию AMPK путем защиты AMPK от дефосфорилирования.
Коснемся слегка интеграции АМФК в метаболическую сеть клетки для осознания сложности всего происходящего (беречь не буду).
Препарат для лечения диабета 2 типа метформин: механизм действия включает активацию AMPK в гепатоцитах. Метформин вызывает энергетический стресс путем ингибирования комплекса I дыхательной цепи в митохондриях. Это приводит к изменению отношения АТФ к АМФ и канонической активации АМФК и фосфорили -рованию ацетил-КоА-карбоксилазы (ACC) в качестве основного участника изменений в синтезе липидов, которые индуцируются метформином. Она, в свою очередь, модулирует чувствительность к инсулину и поглощение глюкозы в мышцах.
Существует 12 возможных комбинаций комплексов αβγ АМФК в зависимости от того, какие субъединицы составляют комплекс. Конкретные композиции субъединиц позволяют различным комплексам AMPK реагировать на различные типы стрессовых стимулов. Также разные субъединицы имеют разную субклеточную локализацию в клетках, например α2 может иметь ядерную локализацию. Но в целом AMPK локализуется в лизосомах благодаря взаимодействию с аксином, белком, который лучше всего характеризуется своей ролью в регуляции пути WNT. В этом контексте аксин также локализуется в лизосомах, где он взаимодействует с киназой печени B1 (LKB1; также известна как STK11, является опухолевым супрессором). LKB1 ответственна за большую часть активации AMPK при энергетическом стрессе.
Комплекс AMPK активируется путем фосфорилирования Thr172 в петле активации каталитической (α) субъединицы с помощью вышестоящей киназы. В ответ на энергетический стресс AMP стимулирует взаимодействие с аксином, что позволяет фосфорилировать Thr172 путем активации LKB1 и AMPK. Это позволит совместно регулировать mTOR, который играет противоположную роль по отношению к AMPK: он стимулирует анаболические пути в условиях высокой питательности. AMPK и mTOR являются компонентами древних консервативных путей, которые развивались как антагонистический механизм, похожий на инь-янь, контролирующий катаболизм и анаболизм.
Отметим, что глюкозное голодание активирует AMPK независимо от канонического AMP-зависимого аллостерического механизма. Скорее всего, связывание гликолитического фермента альдолазы с его субстратом фруктозо-1,6-бисфосфатом (FBP) регулирует образование комплекса AMPK-аксин и активность AMPK.
Учитывая роль пути AMPK-ACC в регуляции синтеза жирных кислот, активация AMPK также является привлекательным вариантом лечения для состояний, связанных с повышенной выработкой жирных кислот, таких как неалкогольная жировая болезнь печени (НАЖБП).
mTOR является ключевой регуляторной киназой, которая играет роль в регуляции рибосомальной трансляции мРНК в белки и важна для роста и выживания клеток. Передача сигналов mTOR усиливается при многих раковых заболеваниях, и поиск эффективных ингибиторов mTOR является основной задачей многих фармацевтических компаний.
mTOR активируется питательными веществами, факторами роста и другими стимулами и интегрирует сигналы от множества восходящих киназ, таких как пути киназы PI3K-Akt и Ras-Raf, и ингибиторов этих путей, таких как PTEN (гомолог фосфатазы и тензина, природный ингибитор PI3K-Akt), которые также ингибируют mTOR. АМФК является важным восходящим регулятором, и активация AMPK с помощью киназы печени B1 (LKB1) или сестрина 2 приводит к фосфорилированию комплекса туберозного склероза (TSC), который ингибирует mTOR.
Химиотерапия, нацеленная на mTOR, включает как прямые ингибиторы, такие как эверолимус и темсиролимус, так и ингибиторы киназ, расположенные выше по течению.
АФК-зависимая активация р53 приводит к индукции двух р53-регулируемых генов, а именно, сестрина 1 и сестрина 2, которые путем активации AMPKα ингибируют передачу сигналов mTOR . АФК (активные формы кислорода) могут сами опосредованно через глутатионовый косплекс активировать AMPKα. Эффекты различных индукторов АФК зависят от клеточного контекста; например, некоторые агенты, такие как куркумин, вызывают аутофагическую гибель клеток в раковых клетках толстой кишки, и предположительно АФК-зависимое ингибирование mTOR куркумином также способствует этому ответу.
Транскрипционные белки Sp1, Sp3 и Sp4 активно экспрессируются в раковых клетках и опухолях, а высокая экспрессия Sp1 в опухолях является негативным прогностическим показателем выживаемости пациентов с раком легких, поджелудочной железы, желудка, глиомы, простаты и молочной железы. Факторы транскрипции Sp играют важную роль в пролиферации, выживании и миграции / инвазии раковых клеток.
АФК-индуцирующие противораковые агенты индуцируют каскад событий, в которых АФК-зависимое эпигенетическое подавление c-Myc вызывает снижение экспрессии c-Myc-регулируемых микроРНК miR-27a и miR-20a / miR-17-5p, что приводит к индукции miR регулируемых транскрипционных репрессоров ZBTB10 / ZBTB34 и ZBTB4 соответственно. Репрессоры ZBTB связывают богатые аминокислотами GC сайты ДНК, чтобы вытеснить Sp1, Sp3 и Sp4. Эти сайты, кстати, особо чувствительно к эпигенетическому метилированию.
Индукторы АФК также могут быть эффективны для комбинированной терапии, поскольку многие Sp-регулируемые гены играют роль в лекарственной и радиационной устойчивости.
Последний раз редактировалось albert52; 10.01.2020 в 03:13..
|